Computational chemistry, computational biology services
Access to cutting-edge computational tools and resources
We help increase your success in:
We are fully committed to the success of your discovery projects
Hit/Lead Optimization
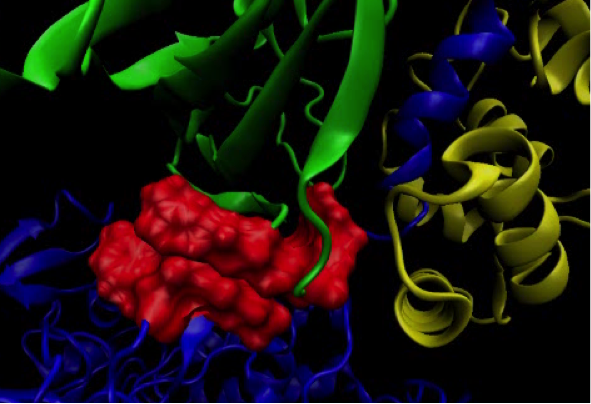
Molecular dynamics simulations
Complementarity of ligand flexibility and protein flexibility
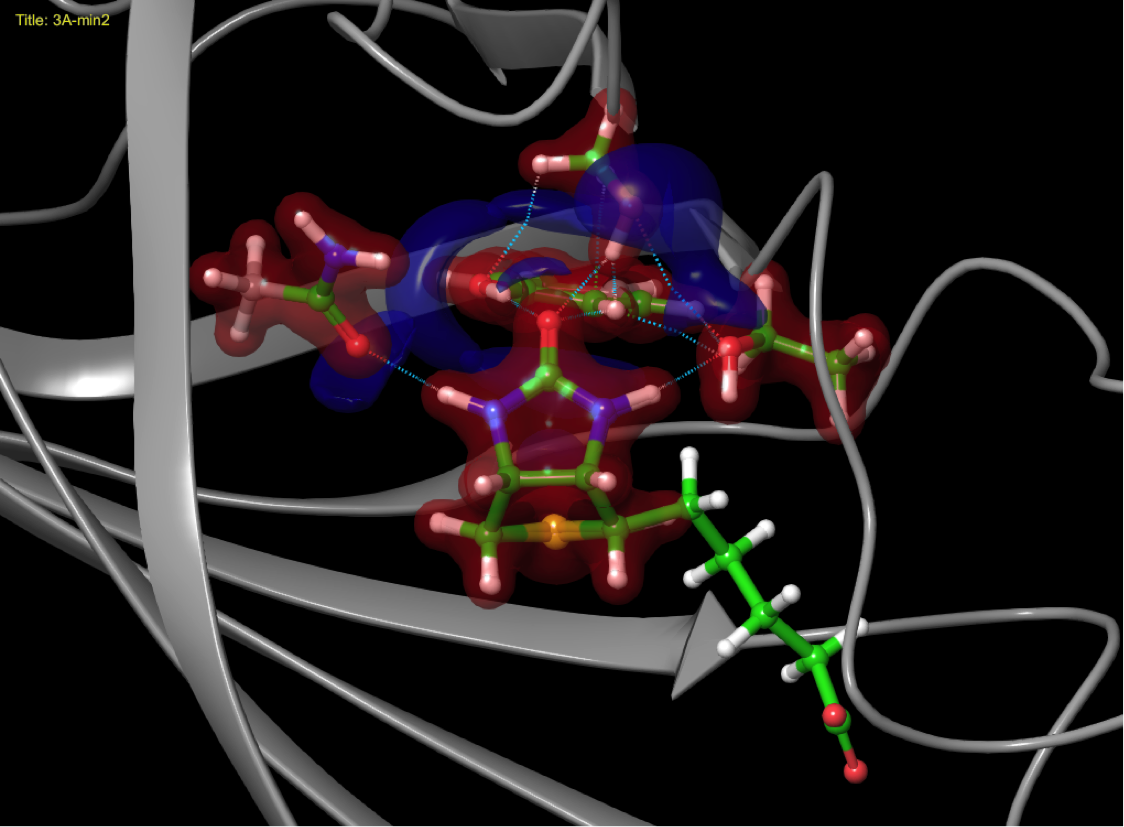
Quantum mechanical simulations
Charge sharing between ligand and biomolecule as it impacts binding
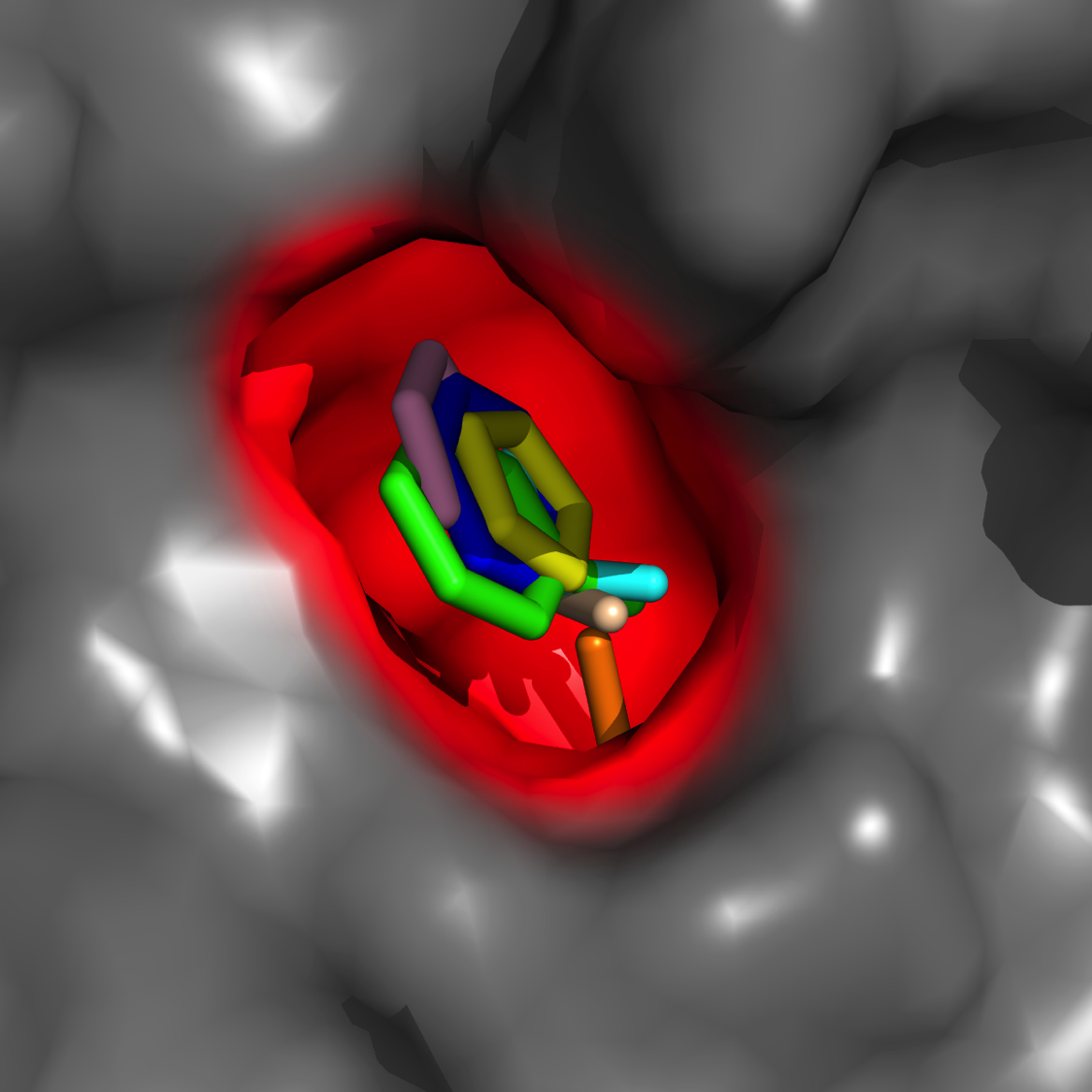
Hot-spot mapping*
Locating novel pockets/sub-pockets of high interaction energy, druggability
*"Diverse Fragment Clustering and Water Exclusion Identify Protein Hot Spots" Kulp et al. JACS 2011 133(28), 10740-10743.
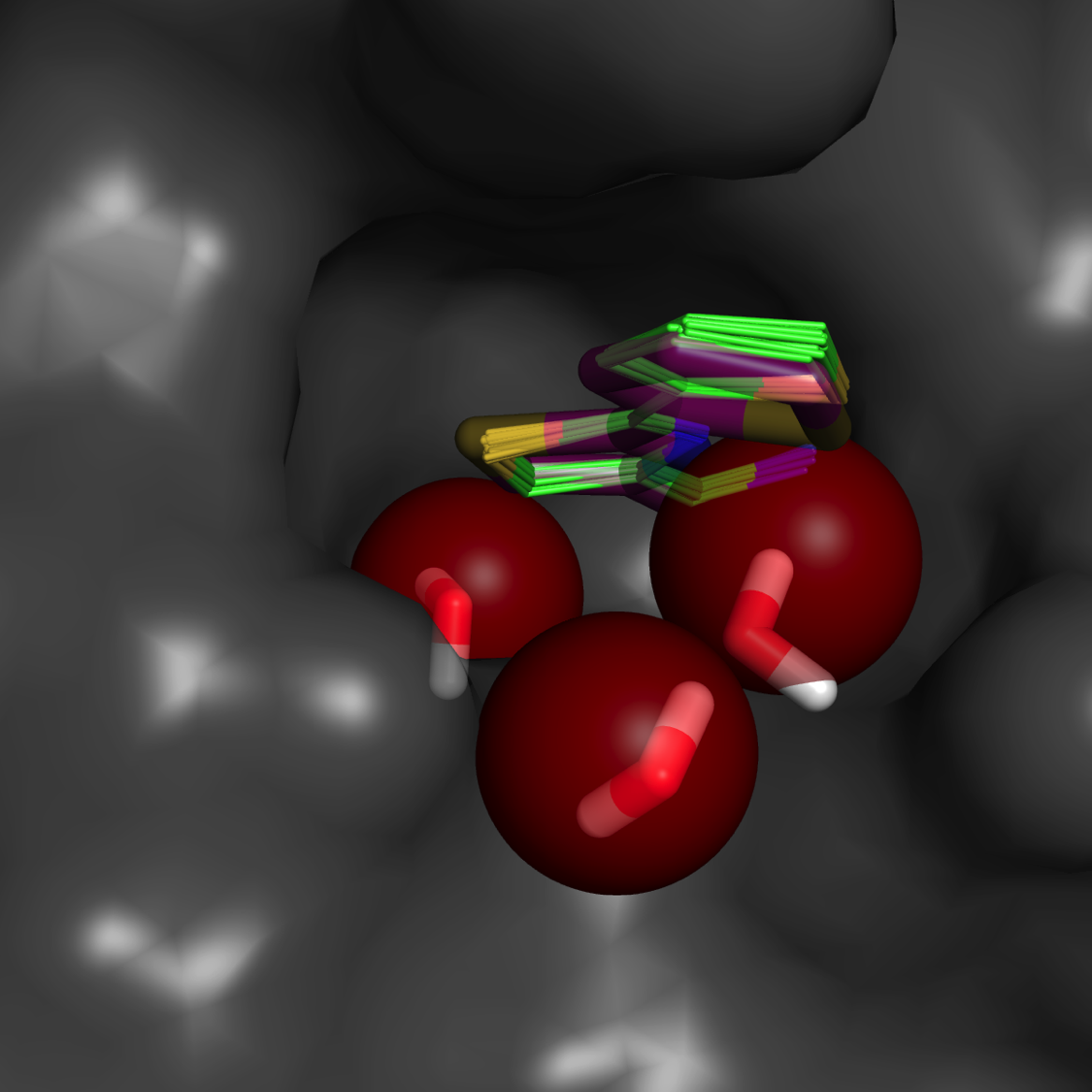
Water mapping**
Estimate of binding affinity (free energy) and rank-ordering
Accurate predictions based on annealing of chemical potential in a Grand Canonical Monte Carlo simulation
**“A fragment-based approach to the SAMPL3 Challenge” Kulp at al. J Comput. Aided Mol Des. 2012 26(5), 583-94.
Computational Chemistry Services
Ligand-based Drug Design
- QSAR, shape-based screening, core hopping, analog-by-catalog similarity searches, conformational analysis
Structure-based Drug Design
- Homology modeling, docking (induced-fit, covalent, QM-polarized), fragment-based ligand growth and optimization
In silico ADME
- Rapid predictions of pharmaceutically relevant properties
Compound libraries
- Commercially-available libraries for virtual screening
- ZINC (~35 million compounds)
- Emolecules (~6 million compounds)
- Exclusive libraries for virtual and experimental screening
- Blumberg Institute’s 85,000 compound library
- Pennsylvania Drug Discovery Institute’s 10,000 compound/reagent library